Will Aromatase Inhibitors Surpress Testosterone
Metrics: Total Views: 0 (Spandidos Publications: | PMC Statistics: )
Metrics: Total PDF Downloads: 0 (Spandidos Publications: | PMC Statistics: )
This article is mentioned in:
Aromatase inhibitor (AI) treatment is first-line systemic treatment for the majority of postmenopausal breast cancer patients with estrogen receptor (ER)-positive primary tumor. Although many patients benefit from treatment, some will develop resistance, and models mimicking acquired resistance will be valuable tools to unravel the resistance mechanisms and to find new treatments and biomarkers. Cell culture models for acquired resistance to the three clinically relevant AIs letrozole, anastrozole and exemestane were developed by selection and expansion of colonies of MCF-7 breast cancer cells surviving long-term AI treatment under conditions where endogenous aromatase-mediated conversion of androgen to estrogen was required for growth. Four cell lines resistant to each of the AIs were established and characterized. Maintenance of ER expression and function was a general finding, but ER loss was seen in one of twelve cell lines. HER receptor expression was increased, in particular EGFR expression in letrozole-resistant cell lines. The AI-resistant cell lines had acquired ability to grow without aromatase-mediated conversion of testosterone to estradiol, but upon withdrawal of AI treatment, testosterone induced minor growth stimulation. Letrozole, exemestane and tamoxifen were able to abrogate the testosterone stimulation but could not reduce growth to below the level in standard growth medium with AI, demonstrating cross-resistance between letrozole, exemestane and tamoxifen. In contrast, fulvestrant totally blocked growth of the AI resistant cell lines both after withdrawal of AI and with AI treatment. These data show that ER is the main driver of growth of the AI-resistant cell lines and indicate ligand-independent activation of ER. Fulvestrant is an efficient treatment option for these AI-resistant breast cancer cells, and the cell lines will be useful tools to disclose the underlying molecular mechanism for resistance to the different AIs.
Introduction
Endocrine therapy is up-front systemic therapy for breast cancer patients with estrogen receptor (ER)-positive tumors. The antiestrogen tamoxifen has been the key endocrine agent since the 1970s and the meta-analysis after 15 years follow-up of 5 years adjuvant tamoxifen therapy revealed reduction in both recurrence risk and death (1). The third-generation aromatase inhibitors (AIs) letrozole, anastrozole and exemestane were introduced in the 1990s and proved superior to tamoxifen both in advanced disease and in the adjuvant setting (2–5). Consequently, the recommended first-line endocrine therapy for postmenopausal women with ER-positive disease is treatment with a third-generation AI (6). Both primary (de novo) and secondary (acquired) resistance occur, limiting the benefit of AI therapy. This emphasizes the need for early identification of resistance and new treatment options for patients with resistant tumors. Clinical data demonstrating improved outcome by combining AI therapy with HER2 targeted therapy or with PI3K/AKT/mTOR inhibitors (7–9) show that these pathways play important roles in AI-resistant cell growth. However, at present there are no tools to select the patients for specific therapy, and the heterogeneity of clinical resistance (10,11) underscores the need for extensive clinical and basic research to disclose the underlying molecular mechanisms for resistance to AIs.
The estrogen supply for postmenopausal breast tumors originates from circulatory uptake and local synthesis in both tumor cells and surrounding tissue, e.g., adipocytes, fibroblasts and inflammatory cells (12–15). The resistance mechanisms to AIs may differ in tumors depending on systemic estrogen supply and tumors in which the carcinoma cells are able to utilize the endogenous aromatase enzyme for estrogen supply. Cell models, mimicking AI resistance in tumors depending on systemically delivered estrogen, have been established from estrogen-responsive breast cancer cells, which have been adapted to grow under long-term estrogen deprivation (LTED) (16–20). LTED cells grow estrogen independently and cell growth has been shown to occur primarily via cross-talk between ER and growth factor receptor signaling pathways, including HER2, IGF-IR and PI3K (21–26). AIs have no effect on growth of LTED cells, whereas antiestrogens, and in particular the ER down modulator fulvestrant, inhibit growth of LTED cells (21,22,27). The importance of local synthesis versus circulatory uptake for supply of estrogen to the cancer cells has been debated and although most breast carcinoma cells express aromatase, most studies point towards a major importance of uptake of circulatory estrogen (13–15). Treatment with AIs results in total suppression of whole body estrogen synthesis, but the cancer cells have the capability to increase local estrogen production. Therefore, a model with endogenous estrogen synthesis is warranted.
Surrogate models with aromatase overexpressing breast cancer cells have been developed by stable introduction of aromatase cDNA (25,28–30). Cell lines and xenografts with acquired resistance to AIs have been established by long-term AI treatment of MCF-7 and T47D cells with exogenous over-expression of aromatase (25,31,32). Genome-wide analyses have revealed that the expression profiles for the MCF-7 sublines resistant to the non-steroidal compounds letrozole and anastrozole were very similar whereas a different profile was observed in cell lines with acquired resistance to the steroidal inhibitor exemestane (31,33). Ligand-independent activation of ER was found in both letrozole and anastrozole-resistant cell lines (31), whereas exemestane appeared to act as a weak agonist in exemestane-resistant cell lines, resulting in e.g., induction of the EGFR ligand amphiregulin and activation of EGFR signaling (33,34). A xenograft model with aromatase overexpressing breast cancer cells has disclosed adaptive changes resulting in activation of alternate signaling pathways due to increased expression of e.g., EGFR, HER2 and IGF-IR (25).
The regulation of the endogenous aromatase gene (CYP19A1) is very complex (12) and may play a key role in AI resistance. We have discovered culture conditions for MCF-7 cells, under which the growth is dependent on conversion of androgen to estrogen via the endogenous aromatase enzyme (35). Treatment with AIs totally suppressed cell growth under these culture conditions (35,36). However, during long-term treatment with AIs, a small subpopulation of the cells survived and slowly resumed growth. From such cells, we have been able to establish AI-resistant cell lines. These cell lines are unique models, which mimic acquired AI resistance in tumors utilizing endogenous aromatase activity to obtain estrogen stimulated cell growth. This report is an initial characterization of our panel of letrozole-, exemestane- and anastrozole-resistant cell lines, presenting the expression of HER receptors, ER and ER-regulated proteins, and growth response to treatment with AIs, tamoxifen and fulvestrant.
Materials and methods
Cell lines and culture conditions
The parental cell line for the AI-resistant cell lines was MCF-7 subline 0.5 (MCF-7/S0.5), which originates from MCF-7 cells from the Human Cell Culture Bank (Mason Research Institute, Rockville, MD, USA) that have been stepwise adapted to grow with 0.5% fetal calf serum (FCS) (37). The MCF-7/S0.5 (MCF-7) cells were maintained at 37°C in humidified air with 5% CO2 in phenol red-free DMEM/F12 medium (Life Technologies, Carlsbad, CA, USA) supplemented with 1% heat inactivated FCS (Life Technologies), 2 mM GlutaMAX™-1 (Life Technologies) and 6 ng/ml insulin (Sigma-Aldrich, St. Louis, MO, USA). In order to obtain growth, which depends on conversion of androgen to estrogen via the endogenous aromatase enzyme, MCF-7 cells were transferred to medium with 10% newborn calf serum (NCS) (Life Technologies) and 10−7 M testosterone (Sigma-Aldrich) as described previously (35). AI-resistant cell lines were established from MCF-7 cells grown in medium with 10% NCS and 10−7 M testosterone. A culture of MCF-7 cells were treated with AI [10−6 M letrozole, 10−7 M anastrozole or 10−7 M exemetane (Selleck Chemicals, Munich, Germany)] for one week, trypsinized and seeded in serial dilutions in 24-well plates. Single colonies were transferred to new wells and gradually expanded in medium with AI. After ~2–3 months, the isolated colonies gave rise to AI-resistant cell lines, which could be grown in AI-containing medium with a weekly split ratio of ~1:25. The MCF-7 cell line was authenticated in January 2014 by DNA profiling using short tandem repeat loci performed by Leibniz-Institut DSMZ (Braunschweig, Germany) and found to be matching the genetic profile reported for the MCF-7 cell line (DSMZ ACC 115).
Growth experiments
For dose-response growth experiments, MCF-7 cells were seeded in 24-well plates (Nunc) in their standard growth medium and after one day, they were switched from 1% FCS to 10% NCS. On day 2, treatment was initiated and renewed on day 5. On day 7, cell number was determined by a crystal violet colorimetric assay (38). One week prior to the dose-response growth experiments, AI and testosterone were withdrawn from the growth medium for resistant cell lines. The AI-resistant cell lines were seeded in medium with 10% NCS, experimental medium added on day 2, renewed on day 5 and cell number determined on day 7 as described above. For combination of AI and antiestrogen treatment, the AI-resistant cell lines were seeded in their standard growth medium and treatment from days 2 to 7 with 10−6 M tamoxifen (Sigma-Aldrich) and 10−7 M fulvestrant (ICI 182.780; Tocris Bioscience, Bristol, UK) was performed in standard growth medium with the respective AI. All growth experiments were performed with four sample replicates or more and repeated at least twice with similar results.
Western blot analysis
Lysates (RIPA buffer; 100 nM NaCl, 20 mM Tris base, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 nM EDTA, pH 8.0) were prepared from early passages (5 and 6) of four cell lines resistant to each of the three AIs; letrozole, anastrozole and exemestane grown in their standard growth medium and from the selected AI-resistant cell lines; LetR-1, LetR-3, ExeR-1 and ExeR-3 grown in standard growth medium in passages between 15–30. Lysates from MCF-7 cells grown in standard growth medium with 1% FCS or grown for 21 weeks with 10% NCS + 10−7 M testosterone were used for comparison. For analysis of estrogen-regulated proteins, lysates were prepared from MCF-7 cells and from AI-resistant cell lines (withdrawn from AI and testosterone for one week) grown for five days with 10% NCS and 10% NCS supplemented with estradiol (Sigma-Aldrich), testosterone and testosterone in combination with AIs. SDS-PAGE page and immunoblotting were performed as described previously (39). Antibodies used against the proteins were: β-actin (A5441) from Sigma-Aldrich); Bcl-2 (M0887), EGFR (M7298), HER2 (A0485) and HER3 (M7297) from Dako (Glostrup, Denmark); ERα (RM-9101), progesterone receptor (PR-A and PR-B) (RM-9102) and Hsp70 (MS-482-PO) from Neomarkers (Fremont, CA, USA) and HER4 (4795) from Cell Signaling Technology (Danvers, MA, USA). Blots were washed four times with TBS/0.1% Tween-20 followed by incubation for 1 h with species-specific peroxidase-conjugated secondary antibodies (Dako). Detection was done using ECLplus reagent (GE Healthcare) and a Fujifilm image reader (LAS1000).
Quantitative RT-PCR
MCF-7 cells were grown with 10% NCS + 10−7 M testosterone for five days and AI-resistant cell lines were grown in their standard medium. RNA was isolated using PureLink Micro-to-Midi Total RNA Purification system (Life Technologies) and reverse transcribed to cDNA by the High-Capacity RNA-to-cDNA kit (Life Technologies). The real-time PCR analysis was performed with Power SYBR® Green PCR Master Mix (Life Technologies) using a Rotor-Gene 3000 (Corbett Life Science, Sidney, Australia). All experiments were conducted in accordance to the manufacturer's recommendations. Results were calculated based on a real-time RT-PCR relative quantification strategy and presented as mean relative gene expression levels, compared to the parental MCF-7 cells. Primers and programs for CYP19A1 were as previously described by Díaz-Cruz et al (40).
Statistical analysis
Two-tailed t-test with Bonferroni adjusted p-values for multiple group comparisons was used. The level of statistical significance was set to p<0.05, and indicated by asterisks in the figures.
Results
Testosterone stimulation of MCF-7 cells
To study the effect of AIs and acquired AI resistance, a model system in which cell growth is stimulated by estradiol produced via aromatase-mediated conversion of testosterone is required. Newborn calf serum (NCS) contains low amount of estrogenic activity and MCF-7 cells require estrogen supplementation to grow continuously in 10% NCS (35). Both estradiol and testosterone exerted dose-dependent growth stimulation of MCF-7 cells in medium with 10% NCS (Fig. 1). Maximal growth stimulation of 13-fold was obtained with estradiol concentrations from 10−11 M (Fig. 1A), whereas maximal stimulation of 8-fold was seen with testosterone in concentrations of 0.1–1.0 μM (Fig. 1B).
Establishment of AI-resistant cell lines and determination of ER, PR, Bcl-2, HER receptors and CYP19A1 mRNA
The testosterone stimulation of MCF-7 cell growth can be completely abrogated by addition of the third-generation AIs, letrozole, anastrozole and exemestane (36), but after long-term treatment colonies of cells grow out. We have selected four cell lines resistant to each of the three AIs, letrozole, anastrozole and exemestane, from isolated single colonies from cultures treated for long-term (≥2 months) with 10−6 M letrozole, 10−7 M anastrozole and 10−7 M exemestane, respectively (see Materials and methods). An initial analysis for expression of ER and the ER-regulated proteins; progesterone receptor (PR-A and PR-B) and Bcl-2 as well as the HER receptors, was performed on the cells harvested after 2.5 months with the respective AI (Fig. 2). All but one AI-resistant cell line maintained ER expression and the level of ER was comparable or higher than in parental MCF-7 grown with 1% FCS. MCF-7 cells grown with 10% NCS + 10−7 M testosterone had very low level of ER (Fig. 3B). PR-B and PR-A were not detectable in the resistant cell lines which were grown continuously in medium with testosterone and AI (Fig. 2). Bcl-2 level was lower in resistant cell lines than in MCF-7 cells grown under standard conditions with 1% FCS. EGFR level was low in MCF-7 cells and also in exemestane-resistant cell lines, whereas increased level of EGFR was seen in all four letrozole-resistant cell lines and in one anastrozole-resistant cell line. Noteworthy, the letrozole-resistant cell line with highest EGFR expression had a very low ER level. HER2 and HER3 were increased in most AI-resistant cell lines compared with MCF-7. In contrast, HER4 was reduced in letrozole- and anastrozole-resistant cell lines compared with parental MCF-7 cells grown in 1% FCS, whereas HER4 was increased in exemestane-resistant cell lines.
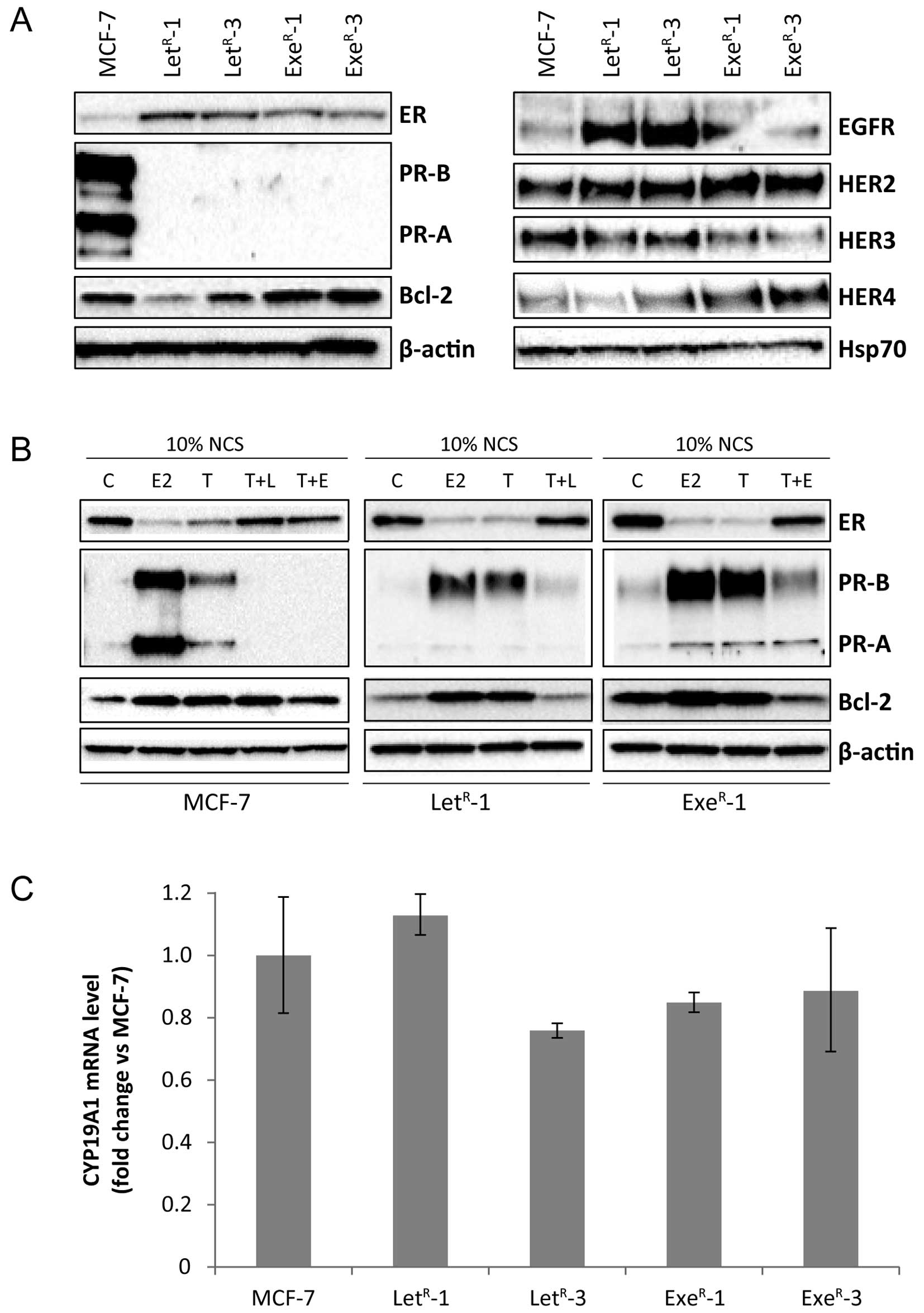 | Figure 3Expression of ER, PR, Bcl-2, HER receptors and CYP19A1 mRNA in MCF-7 and AI-resistant cell lines. (A) Western blot analysis of lysates from MCF-7 cells grown with 10% NCS + 10−7 M testosterone (MCF-7) and LetR-1, LetR-3, ExeR-1 and ExeR-3 grown in their standard growth medium with 10−6 M letrozole and 10−7 M exemestane, respectively. β-actin and Hsp70 were used as loading controls. (B) Western blot analysis of lysates from MCF-7, LetR-1 and ExeR-1 cells grown for five days in 10% NCS (C) or 10% NCS + 10−12 M estradiol (E2), 10% NCS + 10−7 M testosterone (T), 10% NCS + 10−7 M testosterone + 10−6 M letrozole (T + L), 10% NCS + 10−7 M testosterone + 10−7 M exemestane (T+E). LetR-1 and ExeR-1 cells were withdrawn from testosterone and their respective AI one week before onset of experiment. β-actin was used as loading control. (C) CYP19A1 mRNA level in MCF-7 cells grown with 10% NCS + 10−7 M testosterone for five days and AI-resistant cell lines grown in their standard medium determined by quantitative RT-PCR. |
For further analyses, we selected two cell lines resistant to the non-steroidal AI letrozole, LetR-1 and LetR-3, and two cell lines resistant to the steroidal AI exemestane, ExeR-1 and ExeR-3. Initially, we tested the stability regarding expression of ER, PR, Bcl-2 and HER receptors. Fig. 3A is a representative experiment with determination of protein expression in AI-resistant cells from passages in which they have obtained stable growth rates in presence of the respective AI. The resistant cell lines maintained ER expression and the level of ER was higher in both the letrozole- and exemestane-resistant cell lines compared with MCF-7 cells grown with 10% NCS + 10−7 M testosterone. It should be mentioned that the ER content varies with the growth conditions and that ER level was low in MCF-7 cells grown with 10% NCS + testosterone as can be seen in Fig. 2. PR-A and PR-B were expressed at high levels in MCF-7 cells grown with 10% NCS + testosterone and PR continued to be undetectable in the AI-resistant cell lines grown in medium with their respective AI, whereas Bcl-2 level varied between the AI-resistant cell lines (Fig. 3A). EGFR level was significantly higher in letrozole-resistant cell lines compared with MCF-7 and exemestane-resistant cell lines. HER2 level appeared to be slightly higher in resistant cells, whereas HER3 was expressed at level comparable with MCF-7. As also found in the initial analysis (Fig. 2), HER4 expression was higher in exemestane-resistant cell lines compared with MCF-7 and letrozole-resistant cell lines (Fig. 3A). In order to explore whether ER was functional, AI was withdrawn from the AI-resistant cell lines for one week and the cells were treated with estradiol, testosterone or testosterone in combination with AI for five days. Estradiol and testosterone induced expression of PR-A and PR-B as well as Bcl-2 in MCF-7 cells and the two AI-resistant cell lines, LetR-1 and ExeR-1 (Fig. 3B). The ER level was reduced in MCF-7, LetR-1 and ExeR-1 grown with estradiol and with testosterone as expected due to reduced stability of estradiol-bound ER. PR-A and PR-B were hardly detectable in MCF-7 cells grown with 10% NCS and when treated with exemestane and letrozole. PR-A and PR-B were expressed at low level in AI-resistant cells grown with 10% NCS and also expressed at low level in 10% NCS + testosterone and the respective AI. Bcl-2 was expressed at comparable level in control cells with 10% NCS and in cells grown with testosterone and AI (Fig. 3B). These data demonstrate that ER is functional in the AI-resistant cell lines and that the AIs inhibit the conversion of testosterone to estradiol.
Aromatase expression is highly regulated at the transcriptional level (12), and to investigate whether the AI-resistant cell lines displayed aberrant aromatase expression, we measured CYP19A1 mRNA expression using real-time RT-PCR. The analysis revealed similar CYP19A1 mRNA level in MCF-7, LetR-1, LetR-3, ExeR-1 and ExeR-3 cells (Fig. 3C).
AI-resistant cell lines display low degree of androgen responsiveness and cross-resistance between letrozole and exemestane
MCF-7 cells grown with 10% NCS displayed ~6–10-fold stimulation with 10−7 M testosterone, and letrozole and exemestane exerted a dose-dependent growth inhibition with maximum inhibition to the basal level with 10−7 M and 10−6 M AI (Fig. 4). The growth rate of the AI-resistant cell lines increased gradually during the first 15 weeks in medium with AI and then a constant cell population doubling time of ~30 h was achieved (corresponding to a weekly split ratio ~1:25). In comparison, the weekly split ratio for MCF-7 cells grown with 1% FCS is 1:40 (data not shown). To test whether the AI-resistant cell lines maintain the ability to be androgen-stimulated, testosterone and AI were withdrawn for one week from the AI-resistant cell lines before onset of the experiments shown in Fig. 4. Testosterone increased growth of all AI-resistant cell lines, mean stimulation was 2.7-, 2.5-, 1.4- and 3.3-fold for LetR-1, LetR-3, ExeR-1 and ExeR-3, respectively. A dose-dependent inhibition of the testosterone-stimulated cell growth was seen for AI-resistant cell lines treated with letrozole (Fig. 4A and D) and exemestane (Fig. 4B and C). AI treatment of the AI-resistant cell lines abrogated the testosterone-induced cell growth, but did not exert growth arrest as in MCF-7 cells. Noteworthy, the cell number in AI-resistant cell lines grown with 10% NCS was significantly higher than for MCF-7 cells and in contrast to MCF-7 cells, the AI-resistant cell lines could be propagated continuously in medium with 10% NCS (data not shown).
Fulvestrant, but not tamoxifen, exerts complete growth arrest of AI-resistant cell lines
Tamoxifen is a weak estrogen antagonist and in medium with 10% NCS + 10−7 M testosterone, a dose-dependent growth inhibition of MCF-7 cells was seen. However, growth was not completely arrested after five days treatment with 10−6 M tamoxifen (Fig. 5A and B) as it is normally observed with MCF-7 cells grown in their standard medium with 1% FCS (41). Fulvestrant exerted complete growth arrest of MCF-7 cells at concentrations from 10−9 M (Fig. 5C and D). Dose-response growth experiments with antiestrogens were performed with AI-resistant cell lines withdrawn from AI treatment for one week (Fig. 5) and with cells grown with AI (Fig. 6). In AI-resistant cell lines grown with 10% NCS + 10−7 M testosterone and withdrawn from AI treatment for one week, tamoxifen induced a dose-dependent growth inhibition down to the level seen in 10% NCS (Fig. 5A and B). Fulvestrant also inhibited growth of AI-resistant cell lines in a dose-dependent manner, but in contrast to tamoxifen, fulvestrant reduced growth of the AI-resistant cell lines to below the level in NCS medium and down to the level seen in MCF-7 cells treated with 10−7 M fulvestrant (Fig. 5C and D). Tamoxifen treatment of AI-resistant cell lines grown in their standard growth medium with their respective AI had no effect or a stimulatory effect, whereas fulvestrant suppressed growth totally, as seen in MCF-7 cells (Fig. 6). To determine whether AI-resistant cell lines were completely growth arrested with fulvestrant, cultures with LetR-1 and ExeR-1 cells were treated with 10−7 M fulvestrant for one week, split and treated for further one week with fulvestrant. After a total of 14 days of treatment with 10−7 M fulvestrant, no viable LetR-1 or ExeR-1 cells could be detected.
Discussion
Adjuvant treatment with AIs postpones or abrogates development of advanced disease, and patients with advanced disease benefit from treatment. However, most patients with advanced disease will eventually progress, and some patients receiving adjuvant AIs progress during therapy. To find new treatment options and new biomarkers for resistant tumors, it is important to gain knowledge of the molecular mechanisms involved in resistance to AIs. In this report, we describe a new series of breast cancer cell lines with acquired resistance to the clinically relevant AIs letrozole, exemestane and anastrozole, which may be useful models for studies of the resistance mechanisms. The resistant cell lines were developed from MCF-7 cells grown under conditions at which the extracellular estrogen supply via serum was low and growth stimulation was mediated via conversion of testosterone to estradiol by the endogenous aromatase enzyme. Long-term treatment of MCF-7 cells with letrozole, exemestane or anastrozole appeared to inhibit growth completely, but some cells survived treatment and gave rise to outgrowth of colonies, which could be isolated and propagated continuously in presence of the respective AI. Initially, the growth rate was slow, but a gradual increase in growth rate was observed during the first 15 weeks of treatment until constant growth rate was achieved. We believe that the surviving colonies arise from cells with inherent resistance, whereas acquired changes may be responsible for the observed increase in growth rate during long-term propagation.
A switch from ER-driven growth to involvement of HER receptor-driven growth has been described for antiestrogen-resistant breast cancer cells (41–44), and model systems have indicated that resistance to AIs may involve cross-talk between ER and growth factor signaling pathways, and ligand-independent activation of ER (27, 45–47). The initial analysis of expression of ER, estrogen-regulated proteins and HER receptors revealed that ER expression was severely reduced in only one (8%) of twelve AI-resistant cell lines, and a similar low fraction of patients were found to be ER-negative at relapse or progression after AI treatment (11). In general, HER receptor level was higher in AI-resistant cell lines than in parental cells, in particular EGFR was increased in the letrozole-resistant cell lines and HER4 was increased in exemestane-resistant cell lines, pointing to involvement of the HER receptor system in AI resistance. Two letrozole and two exemestane resistant cell lines were selected for further studies and the expression pattern of ER, ER-regulated proteins and HER receptors did not change significantly during longer time propagation of the cell lines. ER was functional with respect to stimulation of expression of PR and Bcl-2, and ER protein expression was reduced in both MCF-7 cells and AI-resistant cell lines grown with estradiol and testosterone. Downregulation of ER expression in MCF-7 cells grown with estradiol and with testosterone has been observed before (35,48) and may be explained by destabilization of the ER protein upon binding to estradiol (49). The testosterone-induced expression of the estrogen-regulated proteins PR-A, PR-B and Bcl-2 in the AI-resistant cell lines verified that AI-resistant cells were able to convert testosterone to estradiol via the aromatase enzyme. AI treatment significantly reduced PR expression in AI-resistant cell lines, but PR expression was not totally blocked as in MCF-7 cells, suggesting that the AIs may not completely block the aromatase activity or that PR may be induced by ligand-independent activation of ER. A similar low PR expression was found in AI-resistant cell lines grown with NCS alone, supporting a ligand-independent activation of ER. Upregulation of aroma-tase gene expression could result in insufficient inhibition of the aromatase enzyme in the AI-resistant cells and explain the transcriptional activity of ER. However, the observed similar level of CYP19A1 mRNA in parental MCF-7 cells and in the four tested AI-resistant cell lines does not support this mechanism of resistance.
Cell number in both the letrozole- and the exemestane-resistant cell lines increased 20–25 times during one week in the standard growth medium with the respective AI, and all resistant cell lines displayed a low degree of responsiveness to growth stimulation with testosterone when AI and testosterone were withdrawn for one week. The ability of the AI-resistant cell lines to grow continuously in presence of AI and also in medium with 10% NCS demonstrates that the cell lines have acquired ability to grow without estrogen stimulation, whereas the observed growth response to testosterone indicates that AI-resistant cell lines have progressed from estrogen-dependent to estrogen-responsive cell growth. Letrozole, exemestane and tamoxifen could abrogate the testosterone-mediated growth stimulation, but could not reduce growth to below the level in standard growth medium with AI. In contrast, fulvestrant inhibited growth of the AI-resistant cell lines to below the level in 10% NCS and down to the level of MCF-7 cells which were completely growth arrested by treatment with fulvestrant. To investigate whether fulvestrant completely blocked growth of AI-resistant cell lines, AI-resistant cells treated for one week with fulvestrant were trypsinized and seeded in new culture flasks, but no growth was observed, demonstrating that fulvestrant exerted complete growth arrest of AI-resistant cell lines and supporting that growth of the AI-resistant cell lines depends primarily on ER. The severe growth inhibition seen with fulvestrant treatment of AI-resistant cell lines in their standard growth medium with AI also supports that ER drives growth of the AI-resistant cell lines. The importance of ER-mediated growth and effect of fulvestrant treatment have also been found in exemestane-resistant cell lines derived from aromatase overexpressing MCF-7 cells (34), in MCF-7 breast tumor xenograft with letrozole-resistant aromatase overexpressing cells (50), and in the LTED model system (26). Clinical benefit rates of 32% were found in two phase III studies with fulvestrant treatment of patients with advanced disease after progression on a non-steroidal AI (51,52), supporting that ER may also be an important driver of metastatic breast cancer cells in patients relapsing from AI therapy.
Letrozole and exemestane abrogated the testosterone-mediated growth of both letrozole- and exemestane-resistant cell lines but could not arrest cell growth, demonstrating cross-resistance between the non-steroidal AI letrozole and the steroidal AI exemestane. This is in contrast to clinical studies in which sequential treatment from a non-steroidal to a steroidal AI or vice versa has resulted in clinical benefit for 30–50% of the patients (53). It should be mentioned that the studies included low number of patients (mean 54) and objective response rates were low, average in 8 studies was 11%. Furthermore, mechanisms explaining lack of cross-resistance have not been documented (53), but lack of effective uptake in tumor tissue of particular compounds has been suggested as a possibility (54) and patients may metabolize the compounds differently. Such mechanisms will not be disclosed in our in vitro cell culture model.
Tamoxifen was able to abrogate the testosterone-mediated growth stimulation of the AI-resistant cell lines but could not reduce growth further, demonstrating that tamoxifen is able to inhibit the estrogen-mediated but not the presumed ligand-independent activation of ER. In line with this, we have recently found that ER is the main driver of growth of tamoxifen-resistant cell lines (41). The multi-targeting kinase inhibitors sorafenib and nilotinib could restore the sensitivity of tamoxifen-resistant cell lines to tamoxifen, indicating that growth of tamoxifen-resistant cells occurs via ligand-independent activation of the ER (55). More direct evidence of the inability of tamoxifen to inhibit ligand-independently activated ER was obtained in a study showing that Aurora kinase A upon phosphorylation of ER renders breast cancer cells less sensitive to treatment with tamoxifen (56). In agreement with this, we have shown that Aurora kinase A plays a major role for growth of tamoxifen-resistant breast cancer cell lines, and that inhibition of Aurora kinase A restores the sensitivity to tamoxifen treatment (57). Whether Aurora kinase A plays a major role by ligand-independent activation of ER in AI resistance is under investigation. It should be mentioned that about one third of patients recurring from tamoxifen treatment benefit from treatment with AI (58,59), showing that different mechanisms for tamoxifen and AI resistance also exist.
In conclusion, this report presents a large series of AI-resistant breast cancer cell lines derived from MCF-7 cells, which can be used as models to unravel the molecular mechanisms for growth of breast cancer in patients who recur after an initial response to AI therapy. We found that the majority of AI-resistant cell lines maintained ER expression and function, whereas HER receptor expression was increased. The complete growth inhibition of the AI-resistant cell lines by treatment with the ER down modulator fulvestrant demonstrates that ER is the main driver of growth of AI-resistant cell lines, supporting the potential of fulvestrant therapy for AI-resistant breast cancer.
Acknowledgements
We thank Birgit Reiter for excellent technical assistance. This study was supported by the Danish Cancer Society, A Race Against Breast Cancer, Danish Cancer Research Foundation, Astrid Thaysen's Grant, Wedell-Wedellsborg's Foundation, and Harboe Foundation.
References
| 1 | Early Breast Cancer Trialists' Collaborative Group. Davies C, Godwin J, et al: Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 378:771–784. 2011. View Article : Google Scholar : PubMed/NCBI |
| 2 | Mouridsen H, Gershanovich M, Sun Y, et al: Superior efficacy of letrozole versus tamoxifen as first-line therapy for postmenopausal women with advanced breast cancer: results of a phase III study of the International Letrozole Breast Cancer Group. J Clin Oncol. 19:2596–2606. 2001.PubMed/NCBI |
| 3 | Nabholtz JM, Buzdar A, Pollak M, et al: Anastrozole is superior to tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: results of a North American multicenter randomized trial. Arimidex Study Group J Clin Oncol. 18:3758–3767. 2000. |
| 4 | Paridaens RJ, Dirix LY, Beex LV, et al: Phase III study comparing exemestane with tamoxifen as first-line hormonal treatment of metastatic breast cancer in postmenopausal women: the European Organisation for Research and Treatment of Cancer Breast Cancer Cooperative Group. J Clin Oncol. 26:4883–4890. 2008. View Article : Google Scholar : PubMed/NCBI |
| 5 | Dowsett M, Cuzick J, Ingle J, et al: Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J Clin Oncol. 28:509–518. 2010. View Article : Google Scholar |
| 6 | Goldhirsch A, Wood WC, Coates AS, et al: Strategies for subtypes - dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. 22:1736–1747. 2011. View Article : Google Scholar : PubMed/NCBI |
| 7 | Schwartzberg LS, Franco SX, Florance A, O'Rourke L, Maltzman J and Johnston S: Lapatinib plus letrozole as first-line therapy for HER-2+ hormone receptor-positive metastatic breast cancer. Oncologist. 15:122–129. 2010. View Article : Google Scholar |
| 8 | Baselga J, Campone M, Piccart M, et al: Everolimus in post-menopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 366:520–529. 2012. View Article : Google Scholar |
| 9 | Burris HA III: Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 71:829–842. 2013. View Article : Google Scholar : PubMed/NCBI |
| 10 | Miller WR: Aromatase inhibitors: prediction of response and nature of resistance. Expert Opin Pharmacother. 11:1873–1887. 2010. View Article : Google Scholar : PubMed/NCBI |
| 11 | Arnedos M, Drury S, Afentakis M, et al: Biomarker changes associated with the development of resistance to aromatase inhibitors (AIs) in estrogen receptor-positive breast cancer. Ann Oncol. 25:605–610. 2014. View Article : Google Scholar : PubMed/NCBI |
| 12 | Bulun SE, Sebastian S, Takayama K, Suzuki T, Sasano H and Shozu M: The human CYP19 (aromatase P450) gene: update on physiologic roles and genomic organization of promoters. J Steroid Biochem Mol Biol. 86:219–224. 2003. View Article : Google Scholar : PubMed/NCBI |
| 13 | Lykkesfeldt AE, Henriksen KL, Rasmussen BB, et al: In situ aromatase expression in primary tumor is associated with estrogen receptor expression but is not predictive of response to endocrine therapy in advanced breast cancer. BMC Cancer. 9:1852009. View Article : Google Scholar : PubMed/NCBI |
| 14 | Haynes BP, Straume AH, Geisler J, et al: Intratumoral estrogen disposition in breast cancer. Clin Cancer Res. 16:1790–1801. 2010. View Article : Google Scholar : PubMed/NCBI |
| 15 | Lonning PE, Haynes BP, Straume AH, et al: Recent data on intratumor estrogens in breast cancer. Steroids. 76:786–791. 2011. View Article : Google Scholar : PubMed/NCBI |
| 16 | Briand P and Lykkesfeldt AE: Long-term cultivation of a human breast cancer cell line, MCF-7, in a chemically defined medium. Effect of estradiol. Anticancer Res. 6:85–90. 1986.PubMed/NCBI |
| 17 | Masamura S, Santner SJ, Heitjan DF and Santen RJ: Estrogen deprivation causes estradiol hypersensitivity in human breast cancer cells. J Clin Endocrinol Metab. 80:2918–2925. 1995.PubMed/NCBI |
| 18 | Chan CM, Martin LA, Johnston SR, Ali S and Dowsett M: Molecular changes associated with the acquisition of oestrogen hypersensitivity in MCF-7 breast cancer cells on long-term oestrogen deprivation. J Steroid Biochem Mol Biol. 81:333–341. 2002. View Article : Google Scholar : PubMed/NCBI |
| 19 | Coutts AS and Murphy LC: Elevated mitogen-activated protein kinase activity in estrogen-nonresponsive human breast cancer cells. Cancer Res. 58:4071–4074. 1998.PubMed/NCBI |
| 20 | Pink JJ, Jiang SY, Fritsch M and Jordan VC: An estrogen-independent MCF-7 breast cancer cell line which contains a novel 80-kilodalton estrogen receptor-related protein. Cancer Res. 55:2583–2590. 1995.PubMed/NCBI |
| 21 | Martin LA, Farmer I, Johnston SR, Ali S, Marshall C and Dowsett M: Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem. 278:30458–30468. 2003. View Article : Google Scholar : PubMed/NCBI |
| 22 | Jensen J, Kitlen JW, Briand P, Labrie F and Lykkesfeldt AE: Effect of antiestrogens and aromatase inhibitor on basal growth of the human breast cancer cell line MCF-7 in serum-free medium. J Steroid Biochem Mol Biol. 84:469–478. 2003. View Article : Google Scholar : PubMed/NCBI |
| 23 | Santen RJ, Song RX, Zhang Z, et al: Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr Relat Cancer. 12(Suppl 1): S61–S73. 2005. View Article : Google Scholar : PubMed/NCBI |
| 24 | Staka CM, Nicholson RI and Gee JM: Acquired resistance to oestrogen deprivation: role for growth factor signalling kinases/oestrogen receptor cross-talk revealed in new MCF-7X model. Endocr Relat Cancer. 12(Suppl 1): S85–S97. 2005. View Article : Google Scholar : PubMed/NCBI |
| 25 | Sabnis G and Brodie A: Adaptive changes results in activation of alternate signaling pathways and resistance to aromatase inhibitor resistance. Mol Cell Endocrinol. 340:142–147. 2011. View Article : Google Scholar |
| 26 | Martin LA, Ghazoui Z, Weigel MT, et al: An in vitro model showing adaptation to long-term oestrogen deprivation highlights the clinical potential for targeting kinase pathways in combination with aromatase inhibition. Steroids. 76:772–776. 2011. View Article : Google Scholar : PubMed/NCBI |
| 27 | Miller TW, Balko JM, Fox EM, et al: ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 1:338–351. 2011. View Article : Google Scholar : PubMed/NCBI |
| 28 | Zhou DJ, Pompon D and Chen SA: Stable expression of human aromatase complementary DNA in mammalian cells: a useful system for aromatase inhibitor screening. Cancer Res. 50:6949–6954. 1990.PubMed/NCBI |
| 29 | Sun XZ, Zhou D and Chen S: Autocrine and paracrine actions of breast tumor aromatase. A three-dimensional cell culture study involving aromatase transfected MCF-7 and T-47D cells. J Steroid Biochem Mol Biol. 63:29–36. 1997. View Article : Google Scholar |
| 30 | Macaulay VM, Nicholls JE, Gledhill J, Rowlands MG, Dowsett M and Ashworth A: Biological effects of stable overexpression of aromatase in human hormone-dependent breast cancer cells. Br J Cancer. 69:77–83. 1994. View Article : Google Scholar : PubMed/NCBI |
| 31 | Masri S, Phung S, Wang X, et al: Genome-wide analysis of aromatase inhibitor-resistant, tamoxifen-resistant, and long-term estrogen-deprived cells reveals a role for estrogen receptor. Cancer Res. 68:4910–4918. 2008. View Article : Google Scholar : PubMed/NCBI |
| 32 | Brodie A, Jelovac D, Sabnis G, Long B, Macedo L and Goloubeva O: Model systems: mechanisms involved in the loss of sensitivity to letrozole. J Steroid Biochem Mol Biol. 95:41–48. 2005. View Article : Google Scholar : PubMed/NCBI |
| 33 | Masri S, Phung S, Wang X and Chen SA: Molecular characterization of aromatase inhibitor-resistant, tamoxifen-resistant and LTEDaro cell lines. J Steroid Biochem Mol Biol. 118:277–282. 2010. View Article : Google Scholar : |
| 34 | Wang X, Masri S, Phung S and Chen SU: The role of amphi-regulin in exemestane-resistant breast cancer cells: evidence of an autocrine loop. Cancer Res. 68:2259–2265. 2008. View Article : Google Scholar : PubMed/NCBI |
| 35 | Sonne-Hansen K and Lykkesfeldt AE: Endogenous aromatization of testosterone results in growth stimulation of the human MCF-7 breast cancer cell line. J Steroid Biochem Mol Biol. 93:25–34. 2005. View Article : Google Scholar : PubMed/NCBI |
| 36 | Lundqvist J, Hansen SK and Lykkesfeldt AE: Vitamin D analog EB1089 inhibits aromatase expression by dissociation of comodulator WSTF from the CYP19A1 promoter-a new regulatory pathway for aromatase. Biochim Biophys Acta. 1833.40–47. 2013. |
| 37 | Briand P and Lykkesfeldt AE: Effect of estrogen and anti-estrogen on the human breast cancer cell-line MCF-7 adapted to growth at low serum concentration. Cancer Res. 44:1114–1119. 1984.PubMed/NCBI |
| 38 | Lundholt BK, Briand P and Lykkesfeldt AE: Growth inhibition and growth stimulation by estradiol of estrogen receptor transfected human breast epithelial cell lines involve different pathways. Breast Cancer Res Treat. 67:199–214. 2001. View Article : Google Scholar : PubMed/NCBI |
| 39 | Larsen MS, Yde CW, Christensen IJ and Lykkesfeldt AE: Carboplatin treatment of antiestrogen-resistant breast cancer cells. Int J Oncol. 41:1863–1870. 2012.PubMed/NCBI |
| 40 | Diaz-Cruz ES, Shapiro CL and Brueggemeier RW: Cyclo-oxygenase inhibitors suppress aromatase expression and activity in breast cancer cells. J Clin Endocrinol Metab. 90:2563–2570. 2005. View Article : Google Scholar |
| 41 | Thrane S, Lykkesfeldt AE, Larsen MS, Sorensen BS and Yde CW: Estrogen receptor alpha is the major driving factor for growth in tamoxifen-resistant breast cancer and supported by HER/ERK signaling. Breast Cancer Res Treat. 139:71–80. 2013. View Article : Google Scholar : PubMed/NCBI |
| 42 | Frogne T, Benjaminsen RV, Sonne-Hansen K, et al: Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat. 114:263–275. 2009. View Article : Google Scholar : |
| 43 | Nicholson RI, Hutcheson IR, Jones HE, et al: Growth factor signalling in endocrine and anti-growth factor resistant breast cancer. Rev Endocr Metab Disord. 8:241–253. 2007. View Article : Google Scholar : PubMed/NCBI |
| 44 | Morrison G, Fu X, Shea M, et al: Therapeutic potential of the dual EGFR/HER2 inhibitor AZD8931 in circumventing endocrine resistance. Breast Cancer Res Treat. 144:263–272. 2014. View Article : Google Scholar : PubMed/NCBI |
| 45 | Weigel MT GZ, Dunbier A, Pancholi S, Dowsett M and Martin LA: Preclinical and clinical studies of estrogen deprivation support the PDGF/Abl pathway as a novel therapeutic target for overcoming endocrine resistance in breast cancer. Breast Cancer Res. 14:R782012. View Article : Google Scholar : PubMed/NCBI |
| 46 | Brodie A, Macedo L and Sabnis G: Aromatase resistance mechanisms in model systems in vivo. J Steroid Biochem Mol Biol. 118:283–287. 2010. View Article : Google Scholar |
| 47 | Liu S, Meng X, Chen H, et al: Targeting tyrosine-kinases and estrogen receptor abrogates resistance to endocrine therapy in breast cancer. Oncotarget. 5:9049–9064. 2014.PubMed/NCBI |
| 48 | Jensen BL, Skouv J, Lundholt BK and Lykkesfeldt AE: Differential regulation of specific genes in MCF-7 and the ICI 182780-resistant cell line MCF-7/182(R)-6. Br J Cancer. 79:386–392. 1999. View Article : Google Scholar : PubMed/NCBI |
| 49 | Dauvois S, Danielian PS, White R and Parker MG: Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc Natl Acad Sci USA. 89:4037–4041. 1992. View Article : Google Scholar : PubMed/NCBI |
| 50 | Long BJ, Jelovac D, Thiantanawat A and Brodie AM: The effect of second-line antiestrogen therapy on breast tumor growth after first-line treatment with the aromatase inhibitor letrozole: long-term studies using the intratumoral aromatase postmenopausal breast cancer model. Clin Cancer Res. 8:2378–2388. 2002.PubMed/NCBI |
| 51 | Chia S, Gradishar W, Mauriac L, et al: Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in post-menopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol. 26:1664–1670. 2008. View Article : Google Scholar : PubMed/NCBI |
| 52 | Johnston SR, Kilburn LS, Ellis P, et al: Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non-steroidal aromatase inhibitors in postmenopausal patients with hormone-receptor-positive locally advanced or metastatic breast cancer (SoFEA): a composite, multicentre, phase 3 randomised trial. Lancet Oncol. 14:989–998. 2013. View Article : Google Scholar : PubMed/NCBI |
| 53 | Miller WR and Larionov AA: Understanding the mechanisms of aromatase inhibitor resistance. Breast Cancer Res. 14:2012012. View Article : Google Scholar : PubMed/NCBI |
| 54 | Lonning PE: Lack of complete cross-resistance between different aromatase inhibitors; a real finding in search for an explanation? Eur J Cancer. 45:527–535. 2009. View Article : Google Scholar |
| 55 | Pedersen AM, Thrane S, Lykkesfeldt AE and Yde CW: Sorafenib and nilotinib resensitize tamoxifen resistant breast cancer cells to tamoxifen treatment via estrogen receptor alpha. Int J Oncol. 45:2167–2175. 2014.PubMed/NCBI |
| 56 | Zheng XQ, Guo JP, Yang H, et al: Aurora-A is a determinant of tamoxifen sensitivity through phosphorylation of ERalpha in breast cancer. Oncogene. 33:4985–4996. 2014. View Article : Google Scholar |
| 57 | Thrane S, Pedersen AM, Thomsen MB, et al: A kinase inhibitor screen identifies Mcl-1 and Aurora kinase A as novel treatment targets in antiestrogen-resistant breast cancer cells. Oncogene. Nov 3–2014.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI |
| 58 | Buzdar AU, Jonat W, Howell A, et al: Anastrozole versus megestrol acetate in the treatment of postmenopausal women with advanced breast carcinoma: results of a survival update based on a combined analysis of data from two mature phase III trials. Arimidex Study Group Cancer. 83:1142–1152. 1998. |
| 59 | Dombernowsky P, Smith I, Falkson G, et al: Letrozole, a new oral aromatase inhibitor for advanced breast cancer: double-blind randomized trial showing a dose effect and improved efficacy and tolerability compared with megestrol acetate. J Clin Oncol. 16:453–461. 1998.PubMed/NCBI |
Will Aromatase Inhibitors Surpress Testosterone
Source: https://www.spandidos-publications.com/10.3892/ijo.2015.2850





0 Komentar